

DeepMind's AlphaFold and the protein folding problem - what you should know

On 30 November 2020, DeepMind -- the best AI lab in the world -- announced that its deep learning system AlphaFold 2 had been recognized as solving the protein folding problem, a grand challenge in biology [1], which leading experts didn’t think would be solved in their lifetimes. This continues a recurring pattern from them of solving grand challenges decades ahead of expert predictions, beginning with AlphaGo for the board game Go, but DeepMind co-founder & CEO Demis Hassabis goes further: he called it “the most significant thing we’ve done, in terms of real-world impact”. Experts agree: Venki Ramakrishnan, Nobel Prize winner and President of the UK’s Royal Society, says it’s “a stunning advance”, and Carlos Rubiera of the Oxford Protein Informatics Group wrote that “the rest of the protein informatics community is experiencing a scientific cliff-hanger, awaiting DeepMind’s paper with more enthusiasm than Cyberpunk 2077”. (Very funny.) DeepMind are touting applications to everything from drug discovery to environmental sustainability, which sounds like a big deal.
In other words, there’s no shortage of hype. But what is the protein folding problem, and why does it matter? Why did it take 50 years to solve? How did AlphaFold solve it -- indeed, what did it actually do? (Equally relevant: what did it not do that everyone thinks it did?) What “real-world problems” can AlphaFold really help solve? And what’s next?
To make sense of all this, it helps to take a couple steps back.
Many of the world’s biggest problems are tied to proteins and what they do, from developing treatments for diseases to finding enzymes that degrade industrial waste. It is an axiom in molecular biology that “structure determines function”: if you know a protein’s 3D conformation, you know what it does [2]. In 1972, Christian Anfinsen postulated (in his Nobel Prize in Chemistry acceptance speech no less!) that “sequence determines structure”: if you know a protein’s amino acid sequence, you know its conformation, and hence its function. Combining these two gives Anfinsen’s dogma:
Sequence → Structure → Function
A quick primer on proteins to ground the abstract claims above. All life on Earth is made up of amino acids (indeed the same set of 20 amino acids), which are molecules containing carbon, hydrogen, oxygen & nitrogen (C, H, O, N), plus other elements in side chains (“R groups”) specific to each amino acid. Here’s an amino acid:

(Source)
{kind=link}
Amino acids link up to form long chains (sequences) called proteins:

(Source)
{kind=link}
There’s a logic to these sequences; they’re not random. The sequences are encoded in DNA (think “blueprint”), and manufactured by RNA following this blueprint. (This means that DNA determines amino acid sequence, so Anfinsen’s dogma in full is really DNA → Sequence → Structure → Function.)
But while they’re synthesized as linear sequences, they don’t stay linear sequences: they fold and bunch up in complex globular shapes (exactly how, we still don’t understand [3]). So it’s useful to talk about different levels of protein structure. The linear sequence of amino acids is called primary structure. It either coils up into tight spirals (alpha-helices) or folds back and forth into wide flat pieces (beta-sheets); this is secondary structure:
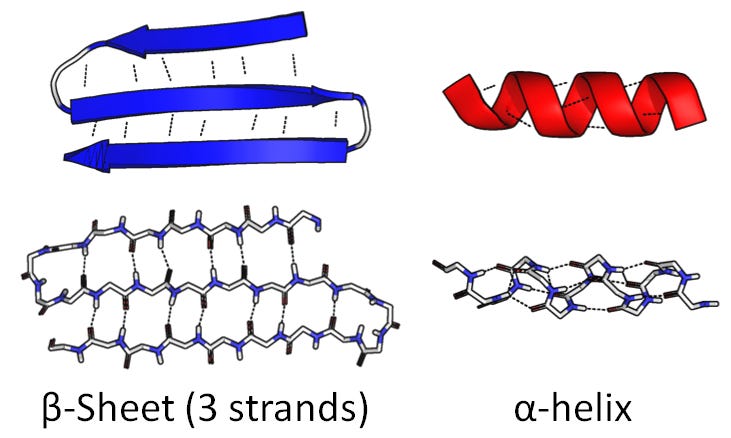
(Source)
{kind=link}
Alpha-helices and beta-sheets fold up to create a protein sequence’s unique tertiary structure, and several sequences can combine to form a quaternary structure comprised of multi-subunit complexes:

(Source)
{kind=link}
While this looks like a mess, there’s (again!) logic to this structure; it’s not random. In particular:
Its physical shape is what gives it a good fit to targets to bind with
Its surface distribution of electric charge matters too
This is why structure → function. (Other physical properties matter too, albeit to a lesser degree.)
For instance, here’s the surface charge distribution of Oryza sativa Lipid Transfer Protein 1:

(Source)
{kind=link}
This 3rd & 4th-level structure is what we want to figure out. Biologists call this protein structure determination. It’s the sequence → structure part of Anfinsen’s dogma.
The issue is that proteins are extremely small, so we can’t just observe their structure directly using an optical (i.e. light) microscope:

(Source)
Instead biologists use two basic approaches to figure out protein structure: measurement & prediction.
As measurement goes, the main workhorse of experimental methods for determining protein structure is x-ray crystallography [4]. In recent years other methods like nuclear magnetic resonance (NMR) and cryogenic electron microscopy (cryo-EM) have seen wider use, but x-ray crystallography remains by far the dominant method:

(Source)
{kind=link}
Now while it looks like we’re trending upward just fine, we’re really not. The issues with these methods are manifold:
They’re hard, expensive, and time-consuming: it takes months to years per structure, using multimillion-dollar equipment
They don’t work for all proteins, e.g. ones embedded in cell membranes (like the ACE2 receptor that SARS-CoV-2 binds to), which are hard to crystallize [5]:

(Source)
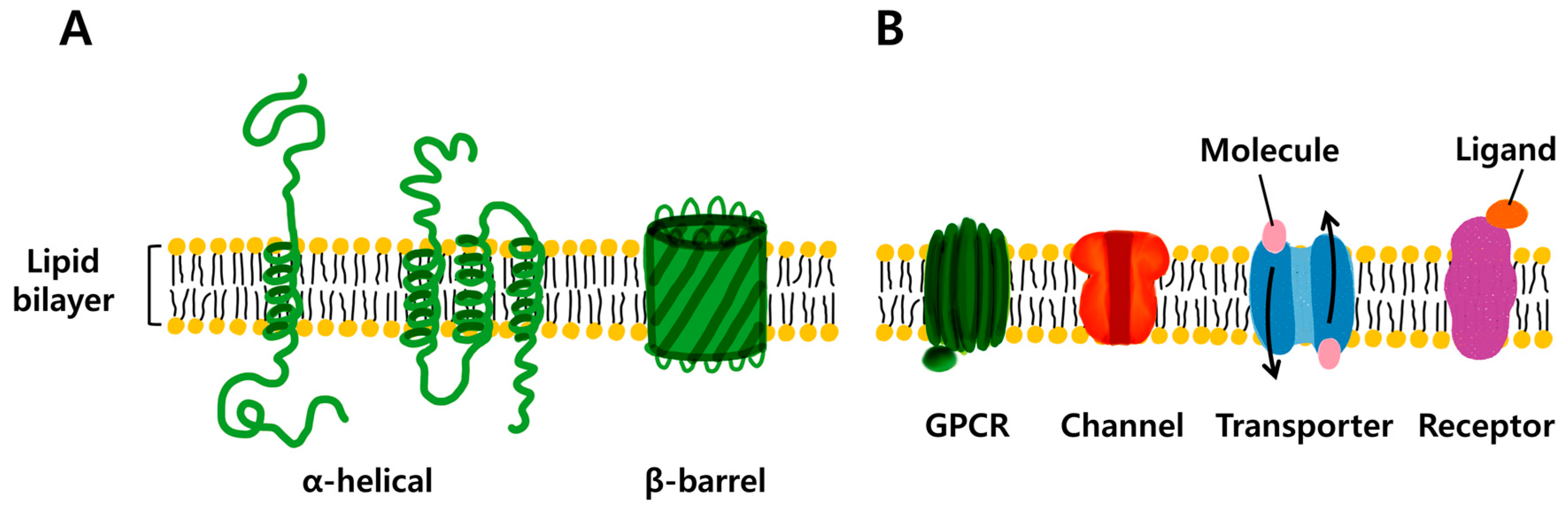{kind=link}
The tremendous effort needed means that we’ve only determined the structure of ~0.1% of the proteins we’ve sequenced -- the Universal Protein database has 180+ million sequences (because the genomics revolution made reading protein sequences cheap & massively scalable -- the DNA → sequence portion of Anfinsen’s dogma), but the Protein Data Bank only has ~170,000 protein structures (sequence → structure). The bottleneck is extreme.
Enter computational prediction. The basic premise is attractive: while you can’t speed up experimental methods by much, you can speed up prediction, and “indefinitely” too; just “add more compute and improve algorithms”. Moore’s law says that compute doubles every 2 years; algorithms get better all the time. Anfinsen’s dogma says predicting structure given sequence should be possible. The sky’s the limit.
Predicting protein structure given amino acid sequence is called the “protein prediction problem”, or colloquially the “protein folding problem”. (As we shall see, there’s a bait-and-switch going on here, but I’ll leave it for the end of this essay.)
So why haven’t we done this already? It’s instructive to think through:
The most obvious way is to directly simulate the physics. This is called molecular dynamics (MD). It involves modeling the forces on each atom in the protein (given its location, charge and chemical bonds with other atoms), calculating each atom’s velocity & acceleration, and evolving the entire simulated protein time-step by time-step, from the beginning when it’s just a linear sequence to its final state as a complex globular structure
The issue is that it’s extremely computationally intensive. A typical protein has thousands of atoms. Since folding is guided by hydrophobic interaction with surrounding water, you need to include water molecules as well, which is tens of thousands more atoms. Since MD involves modeling forces on each atom, you need to consider electrostatic interactions between each atom pair, which goes as the square of the number of atoms, so that’s hundreds of millions of pairs. And you need to run this simulation for billions to trillions of time-steps. All for one protein structure
An alternative way is to find the protein structure that minimizes its potential energy, because stabler configurations have lower energies -- this entails trying out different protein structures and seeing which one has the lowest energy. This makes sense from an evolutionary biology standpoint, because natural selection weeds out all protein sequences except those whose folded state is stable enough and folded fast enough. It’s also relatively easier to simulate, because you can skip all the intermediate time-steps and just figure out the final state. A protein’s energy landscape (vastly simplified) can be visualized like this:
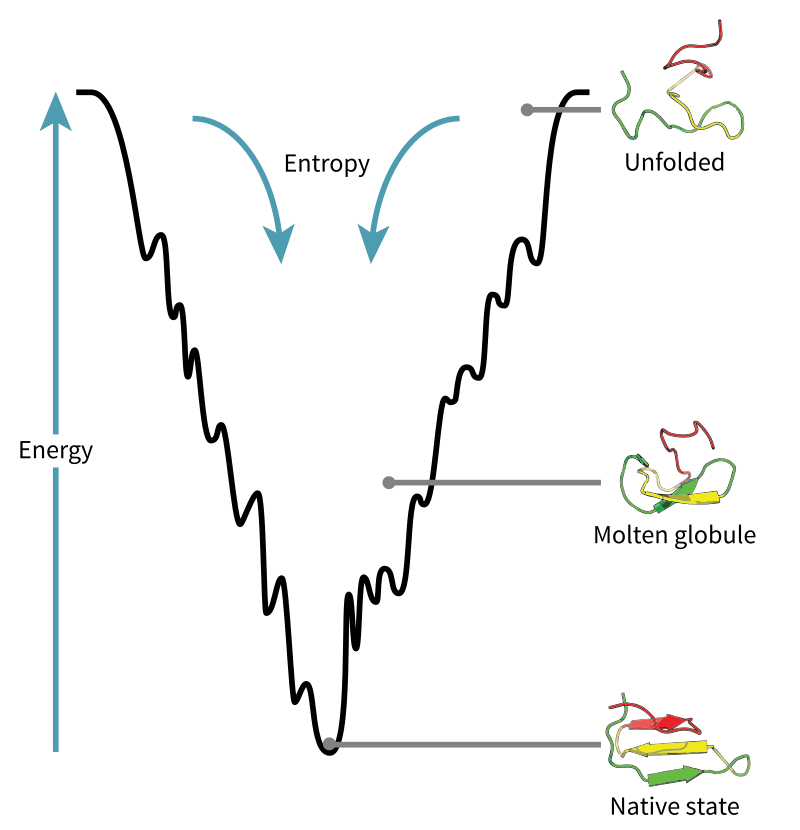
(Source)
{kind=link}
In particular, the energy landscape is a “funnel”, which lets the protein fold to the stable (“native”) state via many possible pathways instead of being restricted to a single one -- natural selection weeds out the latter as they’re more fragile to change. Adding another axis (to make the energy landscape more “landscape”, intuitively) may help:

(Source)
The issue is that it’s still computationally intensive, because “relatively easier to simulate” doesn’t mean actually easy, since the number of possible structures of a single protein is still ridiculously high. We know that protein sequences don’t just randomly try out every possible structure until they find the lowest-energy stable one, because there are so many possibilities that even trying a billion every second would still take them longer than the age of the universe to finish; yet small proteins (<100 amino acids) fold in a single step, and even the large ones typically take just milliseconds. This discrepancy between naïve expectation & reality is called Levinthal’s paradox
At the end of the day we still need to pare down the astronomical number of possible structures for a given protein sequence to find the lowest-energy state it’ll end up in. There have been lots of different attempts:
Specialized hardware: D. E. Shaw Research’s Anton supercomputer runs entirely on ASICs designed especially for molecular dynamics
Distributed computing: Folding@Home
Augmenting computation with human intuition: the University of Washington’s Foldit online puzzle video game
But in the Wild West-like early days, attempts at prediction didn’t do so well. Papers made lofty claims for methods that didn’t stand up to scrutiny when scientists tried applying them to other proteins. Eventually John Moult of the University of Maryland and Krzysztof Fidelis at UC Davis got sick of this, so they started the biannual Critical Assessment of Structure Prediction (CASP) competition to bring more rigor to these efforts, by challenging teams to predict structures of proteins that have been measured via experiment, but haven’t been made public. The predictions are then assessed by a team of independent scientists who don’t know who is making what prediction, making CASP a blind assessment. It’s both competition and info-sharing session: after results are announced, teams present their methods in enough detail that others can replicate & build upon them, so the entire field advances.
The goal is to get a median score of 90 on the Global Distance Test (GDT) metric, which is (roughly speaking) the percentage of amino acids within some max threshold distance from the correct position [6] -- even more informally, the “fraction of the protein correctly predicted”. Why 90 GDT? Because that’s informally considered to be competitive with results from experimental methods. Since large complex proteins have harder-to-predict structure than small simple ones, models are ranked by their median score. To give a sense of what GDT scores correspond to:
Random predictions give GDT 20 (so if your model gets <20 it’s literally worse than useless)
Getting the gross topology right is ~50
Accurate topology is ~70
Getting all the details right to experiment-level precision is >90
It turns out it’s just really hard to accurately predict how a wide variety of proteins fold. Forget about 90 GDT; despite having the best researchers in the world join every contest, the top scorers could barely crack 40 GDT...

...until DeepMind joined the fray, and just blew everyone out of the water with AlphaFold’s 68.5 GDT...

...before improving by an an even bigger margin with AlphaFold 2’s 92.4 GDT:
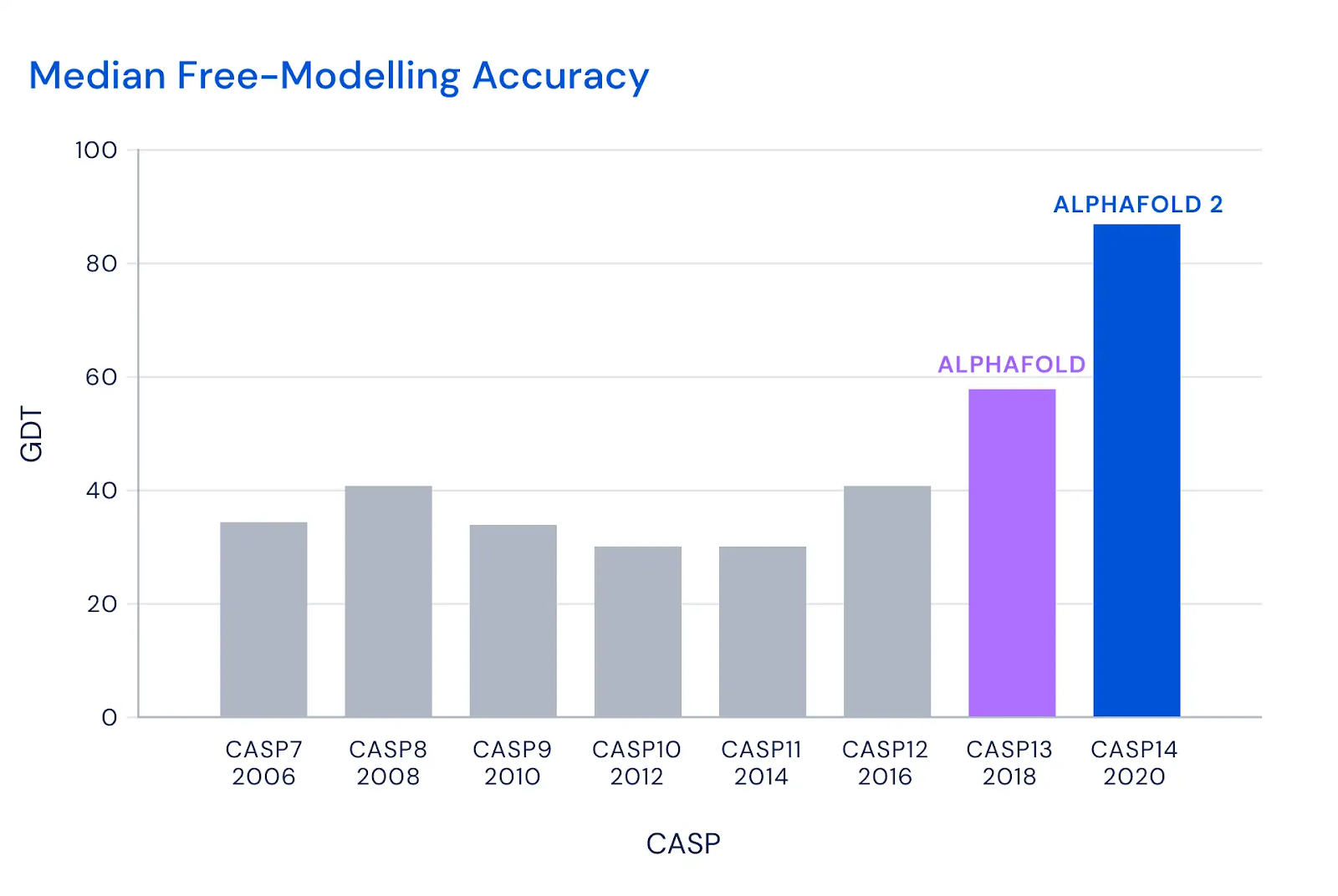
(Source)
AlphaFold 2’s median GDT score across all proteins was 92.4, competitive with results from experimental methods. Even more impressively, AlphaFold 2’s “GDT score dropoff” as protein structure gets more complicated is far lower than that of other models; in other words, while other models do far worse on the most difficult protein structures (close to useless in fact), AlphaFold 2 does nearly as well as it does simple proteins at 87 GDT -- complexity doesn’t faze it:

(Source)
Three remarks:
It’s a misconception (albeit a common one in many pop sci articles) that a 92.4 GDT median score means that AlphaFold 2 predicted 92.4% of protein structures accurately; this is a misunderstanding of what the Global Distance Test metric measures. What it means (informally) is that half of the protein structures predicted got >92.4% of their atoms in approximately the right place
Look at the improvement in the 2nd-last graph above from 2016 (CASP12) to 2020 (CASP14) for other competitors: the best non-DeepMind teams (David Baker’s group at U of Washington & Yang Zhang’s group at U of Michigan) did much better at predicting the hardest protein structures than last time. They improved so much by incorporating many of the ideas from AlphaFold 1
Some experts wondered whether AlphaFold 2’s performance might have been due to “an easier-than-usual crop of protein targets”. They’re wrong -- CASP assessors have concluded that the protein targets for CASP14 2020 were in fact the hardest to date:

(Source)
{kind=link}
We can zoom into AlphaFold 2’s performance a bit more. “g427” is short for “group 427”, the name under which AlphaFold 2’s predictions arrived for blind assessment. As the graph below shows, not only did half of the predicted protein structures score 92.4 or better, 63% of them scored 90 or better (where prediction becomes competitive with experimental measurement) and 88% scored 80 or better. In fact, only 5 out of 93 predictions had GDT <70, i.e. <10% didn’t get the details right:

(Source)
Historically speaking, it’s very rare for one team’s method to dominate others so thoroughly; usually it’ll do better at some proteins and worse at others versus comparably good methods. The Baker and Zhang groups’ methods (2nd & 3rd-best respectively), for instance, run neck and neck. The graph below shows how well Baker did vs Zhang for every single protein structure predicted:

(Source)
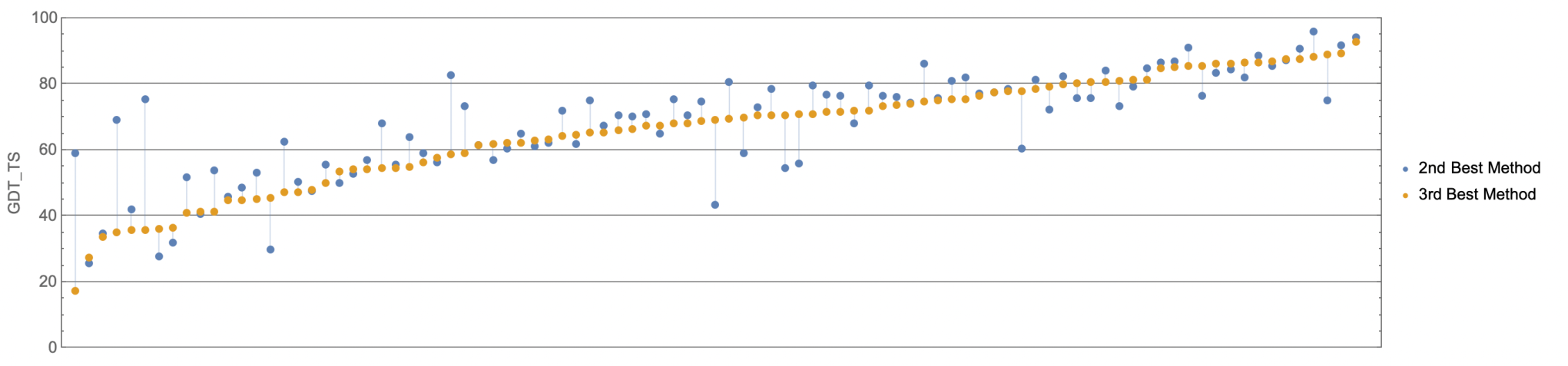{kind=link}
Now here’s AlphaFold 2 versus Baker. The dominance is obscene. There are protein structures for which Baker predicts literal nonsense (at GDT ~20) but AlphaFold 2 predicts at almost experiment-level accuracy (nearly GDT 90):

(Source)
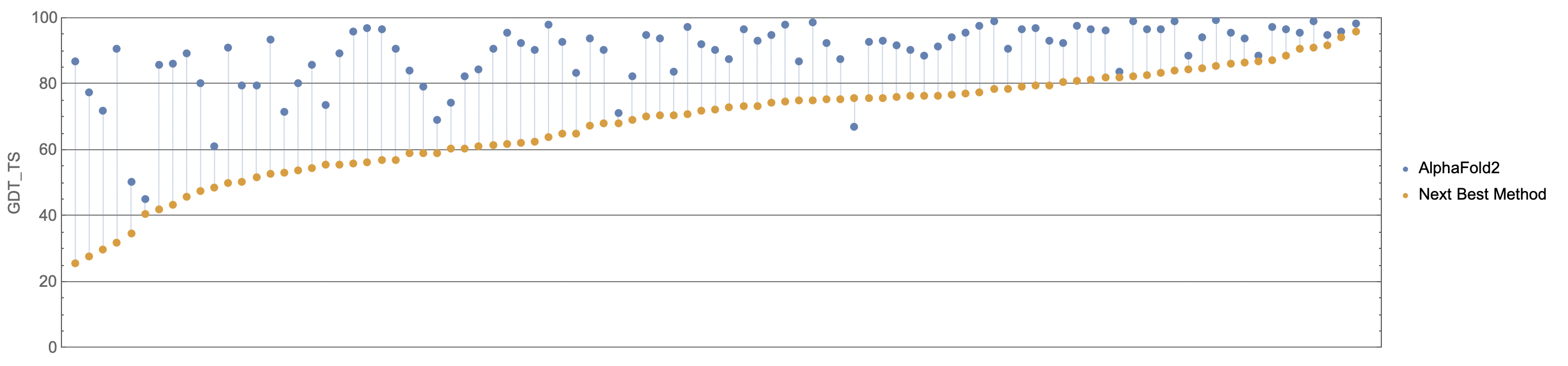{kind=link}
But it gets better. AlphaFold 2 did so well that CASP organizers started worrying DeepMind was cheating. So CASP judge Andrei Lupas gave it a special challenge: predict the structure of a certain membrane protein his research group had been stuck on for ten years. Knowing the structure would help them understand how signals are transmitted across cell membranes. They had the x-ray crystallography data; they just couldn’t interpret it, and they’d tried “every trick in the book”.
No trouble for AlphaFold at all: it predicted a three-part protein, two long helical arms in the middle. Using a technique called molecular replacement, which piggybacks upon a previously-solved protein structure (in this case by AlphaFold) similar to the unknown one to interpret the x-ray crystallography diffraction pattern, Lupas & colleagues figured out the x-ray data: a near-perfect match.
Lupas was flabbergasted:
They could not possibly have cheated on this. I don’t know how they do it. ...This will change medicine. It will change research. It will change bioengineering. It will change everything.
(Given the context, you’ll perhaps forgive his wild enthusiasm.)

(Source)
But it gets even better. AlphaFold 2’s predictions were so good that they sometimes defied experiment results -- even when assessors thought it failed, it got it right. Osnat Herzberg’s group at U of Maryland were studying a protein from the tail of a bacteriophage, a virus that parasitizes bacteria by infecting them and taking over their internal machinery to reproduce -- something like this:

(Source)
AlphaFold 2’s prediction agreed very well with their structure, except for one cis-proline assignment. After reviewing the analysis, they realized the mistake was not AlphaFold’s but theirs:
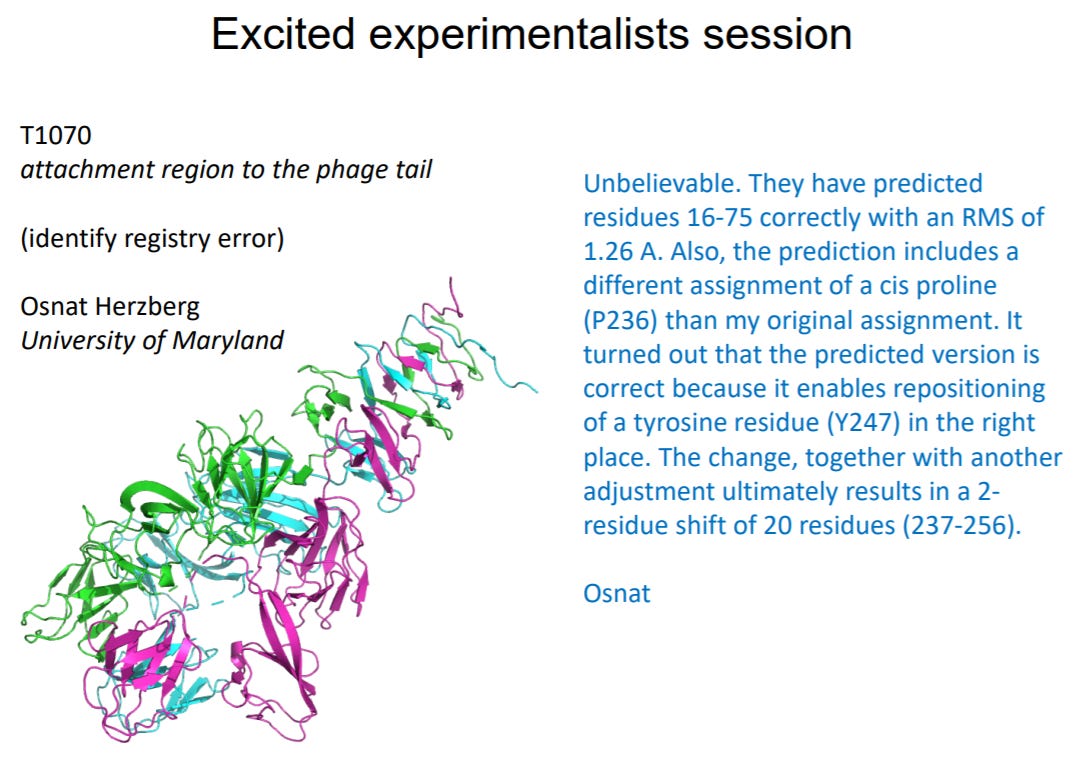
(Source)
So how did DeepMind do it? In short:
Novel approach (completely different from AlphaFold 1’s, which was similar to existing best approaches except plus deep learning)
Lots of compute for training (albeit relatively modest vs most state-of-the-art ML models) and prediction (“inference” -- much more than experts expected [8])
DeepMind’s org structure is totally different from most academic groups, which lets it have a “fast & focused” research paradigm leveraging a level of coordination not available to academics because it’s disincentivized [9]. (This doesn’t mean DeepMind’s approach is better outright -- it’s great for answering questions, not asking them [10].)
The vast amount of data, techniques, & software tools developed by researchers over the decades prior, which DeepMind used to the fullest [11] (although it’s still true that DM did it decades before everyone expected)
A bit more on AlphaFold 2’s novel approach. (Not a lot more unfortunately, as DeepMind wasn’t as generous with AlphaFold 2’s details as they were with AF1 back in 2018, to the consternation of some CASP participants [7], so a high-level sketch will have to do.)
AlphaFold 2 is an attention-based deep learning neural network. Quick primer:
Artificial neural networks (ANNs) are loosely inspired by biological NNs that comprise animal brains. They consist of a bunch of artificial neurons connected together. Connections let neurons transmit signals (numbers) to each other; neurons process signals (i.e. compute some nonlinear sum of the inputs). Both neurons and connections have “weights” (also numbers) that increase or decrease signal strength. These weights are adjusted as the NN “learns”; in effect, learning is adjusting weights

(Source)
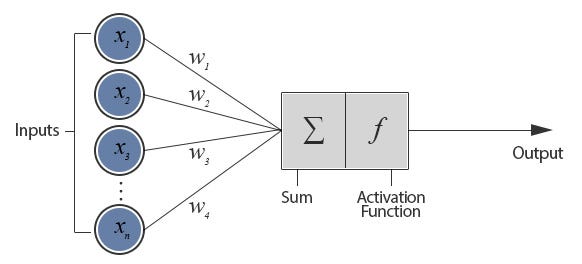{kind=link}
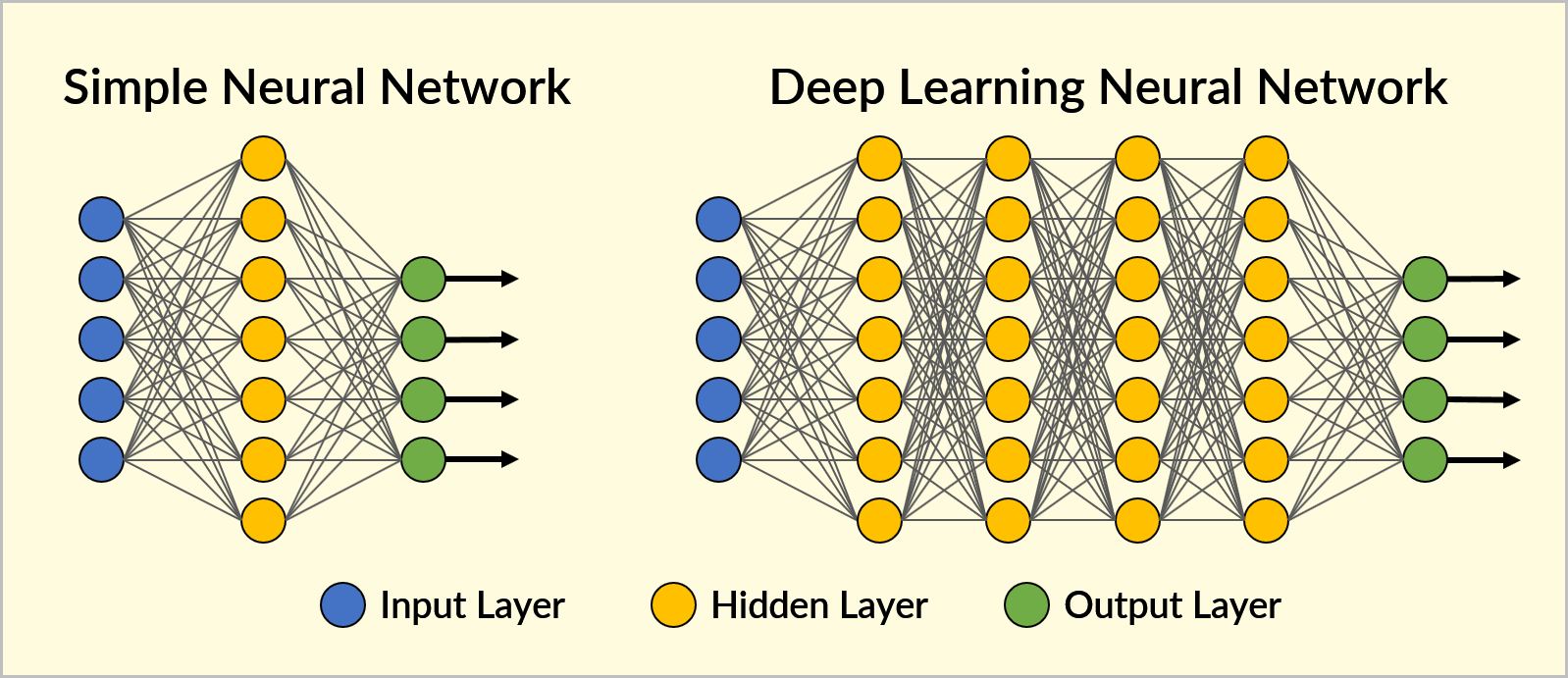
Neurons in NNs are usually aggregated into layers. Different layers may process input signals differently. Here’s a 3-layer NN, with one input layer, one hidden layer and one output layer:

(Source)
{kind=link}
A deep learning NN is just an NN with many hidden layers (sometimes hundreds):

(Source)
{kind=link}
Having many hidden layers lets NNs learn higher-order abstractions, e.g. pixels → edges → edge arrangements → nose / eyes → human face:

(Source)
{kind=link}
Attention (in machine learning) is a technique that mimics the effect of biological attention, by having the AI algorithm identify parts of a larger problem and then piecing them together to get the overall solution (think of it like assembling a jigsaw: piece together local chunks first, then fit these into a whole)
AlphaFold 2’s main architecture looks like this:

(Source)
This is much too vague of course. DeepMind wasn’t much more forthcoming about details, both in their post as well as their CASP14 presentation, which has leftexpertsguessing; all they’ve said is that they’re “preparing a paper on our system to submit to a peer-reviewed journal in due course”.
There is one interesting thing to say about AlphaFold 2’s architecture. Unlike AlphaFold 1 and everyone else, which have some physics knowledge “baked in”, AF2 doesn’t bake in anything. It just learns from the data:

(Source)
This is noteworthy because it continues an ongoing trend of DeepMind’s:
Do task X
Do task X, but with simpler architecture / fewer assumptions baked in
For instance, they did it for board games:

(Source)
{kind=link}
My non-expert take: minimizing baked-in assumptions for your AI architecture is directionally correct if the goal is to build a truly general learner, instead of a specialist whose competence doesn’t transfer to other domains; this is indeed what Demis Hassabis and colleagues at DeepMind aim for.
So it does look like DeepMind did great. But what’s AlphaFold good for, given that the reason CEO Demis Hassabis considers AlphaFold 2’s CASP14 triumph “the most significant thing we’ve done” is its “real-world impact”? Taking a closer look at the list of potential applications reveals a more mixed picture that counters the hype. I can find two applications so far:
Giving computational biologists (like CASP judge Andrei Lupas above) a tool in their kit to help figure out protein structures much faster, in days instead of months/years, if they have the compute / money. This is a big ‘if’. To wit: DeepMind says they trained AlphaFold 2 using 128 TPUv3 cores (comparable to 100-200 GPUs); predicting a single protein’s structure would cost an estimated $10,000 in cloud compute, and renting 128 TPUv2 cores (not v3!) costs half a million a year. The best non-DeepMind teams, Baker et al & Zhang et al, only used 4 GPUs to train their models, which should tell you what kind of resource constraints even top-flight academics have to work with. DeepMind is still figuring out how to “best provide broader access to AlphaFold in a scalable way”
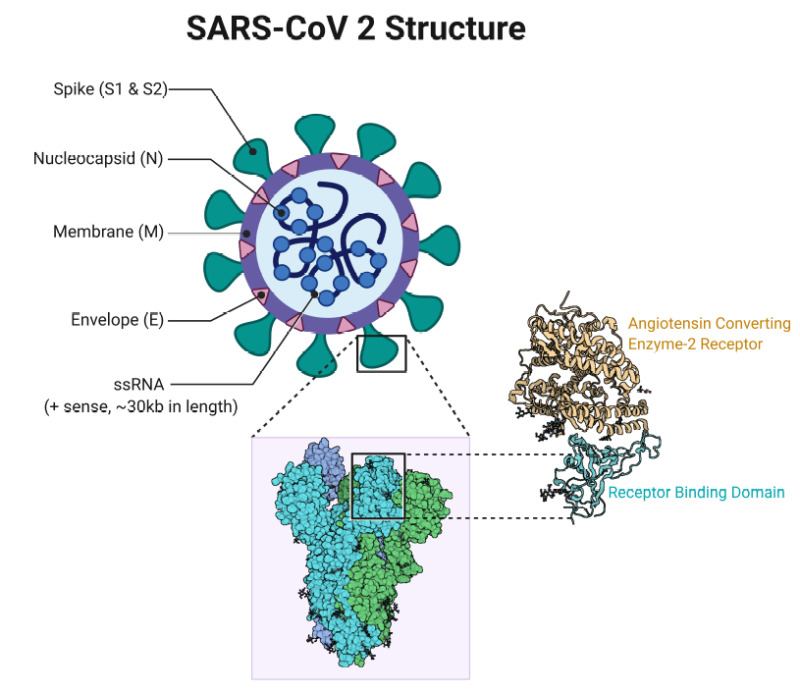
Helping future pandemic response efforts by accelerating vaccine development. AlphaFold 2 predicted several protein structures of the SARS-CoV-2 virus that were previously unknown (e.g. ORF3a), and later proven correct by experimentalists. (Informally: SARS-CoV-2 has “spike” proteins on its surface which it uses to invade human cells, as in the diagram below. If you think of these proteins as “locks”, knowing their structure helps us design “keys” i.e. vaccines to disrupt it. This is called structure based drug design.) But Stephen Curry, a structural biologist at Imperial College London (not to be confused with the NBA player), notes that we’re not quite there yet: getting reliable insights into drug design requires confidence in atomic positions to within a margin of ~0.03 nm, 5x more accurate than AlphaFold 2’s current median performance. Mohammed AlQuraishi, a computational biologist at Columbia University and longtime CASP participant, agrees and adds that even best-case scenario AlphaFold only accelerates the early stage of the drug development pipeline; since most of the cost is in the later stages, the dynamics of drug discovery as practiced today won’t be fundamentally altered

(Source)
{kind=link}
What other potential applications are there?
Study malaria, sleeping sickness, & leishmaniasis, tropical diseases linked to lots of unknown protein structures
Help make sense of gene variations between people that cause disease, which AlQuraishi calls “variant prediction”. To elaborate: proteins mutate, altering their functions/roles, causing diseases. Exactly how mutations alter protein function is a hard problem, so hard that it requires extraordinary statistical power to figure out except for the simplest diseases. Knowing protein structure won’t solve this problem, but it will give more powerful tools to help. DeepMind plans to do exactly this, by working with “small specialist groups” focused on particular diseases
Use novel ways to design & engineer proteins from scratch (“de novo design”): the ability to figure out protein structures in days (rather than months to years) lets scientists invert the design process, by which they first define a “scaffold” (like an enzyme’s active center) and then fine-tune a protein sequence to adopt the scaffold
Accelerating progress in synthetic biology, which is in theory about reengineering living organisms to have new abilities for useful purposes, but is in practice hamstrung by its focus on DNA-based circuitry (more predictable, but trial-and-error and much narrower potential), via protein design from scratch
Help environmental sustainability efforts: being able to synthesize proteins with desired structure can speed development of enzymes that make biofuels and degrade waste plastic
Instead of designing drugs (which requires ~0.03 nm resolution per above), designing protein-based therapeutics (e.g. antibodies & peptides) which don’t need ultra-high resolution
Accelerate progress in the evolutionary analysis of proteins: there’s no shortage of genome sequence data, the bottleneck so far has been figuring out structure (Anfinsen’s sequence → structure → function), which AlphaFold alleviates
Explore the 99.9% of protein sequences in the Universal Protein database whose structures we don’t know yet, to discover new proteins with “exciting functions”
A more “moonshot”-type potential application, once ultra-high-res is achieved, is to design drugs for their poly-pharmacology. As practiced today, conventional medicinal chemistry is about managing downside: make highly selective small molecules that act on one protein at a time, and minimize off-targets. We can instead imagine designing drugs that modulate multiple protein targets (indeed entire signaling pathways) at once. This is pretty ambitious, because signaling pathways get complicated fast. For instance, here’s what it looks like for oxytocin, the hormone released into the bloodstream in response to love and in labor that’s associated with empathy, trust, social bonding and sex:

(Source)
{kind=link}
Again I want to check the hype a bit: AlphaFold won’t revolutionize things as a standalone tool in the short run (CASP judge Andrei Lupas’ breathless proclamations about it changing “medicine, research, everything” aside), but it will definitely be a very useful tool to help in a lot of different areas in the long run.
Which is not to say that it hasn’t gotten some pushback. Some of the more relevant ones:
This isn’t really DeepMind’s fault (they’re just working with available data), but AlphaFold was trained on all 170,000 structures in the Protein Data Bank. We know (from the experimental methods graph above) that this consists overwhelmingly of proteins whose structures were determined via x-ray crystallography. This raises 2 issues: (1) the set of crystallizable proteins isn’t necessarily representative of all proteins; (2) frozen protein structures in crystals may differ from their dynamic structures in cells (“the cytoplasm is not a crystal!”). So what AlphaFold is really predicting isn’t “stable minimal energy state” protein structure, but crystallized protein structure. This may be the reason AlphaFold struggled with predicting structures obtained by NMR instead of x-ray crystallography
What DeepMind solved wasn’t protein folding, but protein prediction, and the distinction matters. (This is the bait-and-switch I mentioned above.) CASP cofounder John Moult knew this, so he was careful in his wording when declaring AlphaFold’s achievement:

(Source)
What’s the difference? Recall how protein folding works: you start with a linear sequence of amino acids, which folds and bunches up due to hydrophobic collapse and other forces, and end up with a globular structure. For complex proteins, this process involves lots of steps. Protein prediction skips all the intermediate steps; protein folding doesn’t:

(Source)
{kind=link}
Why does it matter? Because understanding protein folding helps understand protein misfolding. This is precisely the problem that Folding@Home, the distributed computing project, works on. Protein misfolding usually creates inactive (i.e. harmless) proteins, but sometimes it creates harmful ones. For instance, some neurodegenerative diseases are believed to result from the accumulation of amyloid fibrils formed by misfolded proteins. Many allergies are also caused by protein misfolding, since the immune system doesn’t produce antibodies for certain structures
DeepMind had access to way more compute, and specialized hardware. (“They had more compute” is a perennial gripe in AI research.) This gave them 2 key advantages: rapid prototyping and testing of ideas (weeks → hours), and ideas for NN architectures that wouldn’t even be considered given lack of appropriate hardware (like TPUs). My non-expert take: this isn’t really a strike against DeepMind, but the trend of getting “priced out of the running” does seem to point to a Big Science future for computational science (not just comp bio in particular) where, instead of a “let a thousand flowers bloom” approach, researchers will have to pool their resources in massive international consortiums that purchase hardware at scale, like how high-energy particle physicists have been doing for decades to fund multibillion-dollar projects like CERN
So what’s next? There are several ways in which AlphaFold can still improve, aside from the above concerns:
It’s really slow. AlphaFold takes days to make predictions; AlQuraishi’s method, which uses an algorithm called recurrent geometric networks (RGN), takes milliseconds. (He even made his code publicly available on GitHub.) That’s millions of times faster, albeit less accurate. In some applications, speed matters more
It struggles with protein complexes, i.e. quaternary structures composed of multiple sequences; it’s only good at tertiary structures (single sequences). This matters because lots of important chemical receptors in our body are protein complexes. What makes protein complexes even harder to predict is that their structure can change: for instance, a “closed-tunnel”-shaped protein can open when the right chemical “docks” on its surface, like in the illustration below (this process is key to how brains work). Remember how AlphaFold 2’s predictions scored <70 on 5 out of 93 proteins, i.e. it didn’t get the topology right? Those 5 were all protein complexes

(Source)
{kind=link}
All the caveats and de-hyping aside, I do think that DeepMind did something amazing with AlphaFold 2. I agree with this take by Jack Clark, Strategy & Comms Director @ OpenAI:
AlphaFold is one of the purest examples of why ML-based function approximation is powerful – here’s a system where, given sufficient computation and a clever enough architecture, humans can use it to predict eerily accurate things about the fundamental structure of the biomachines that underpin life itself. This is profound and points to a future where many of our most fundamental questions get explored (or even answered) by dumping compute into a system that can learn to approximate a far richer underlying ‘natural’ process.
What a future, huh?
Footnotes
[1] Nitpick: the thing about “grand challenge” problems is that it depends who you ask, and at what level of granularity. The official Grand Challenges of biology are much grander in scope than protein folding, which doesn’t feature in the list:
Understand the human brain
Understand how the Earth, its climate, and the biosphere interact
Understand biodiversity -- how it influences ecosystem functioning, and how human activities impact it
Synthesize lifelike systems -- to understand the structures/capabilities/processes forming the basis for living systems
Predict individual organisms’ characteristics from their DNA sequence -- to understand how much individual variation within the same species arises from the genome vs gene-environment interactions (“nature vs nurture”)
Every single pop sci article on AlphaFold says protein folding is a “grand challenge” but doesn’t link to an actual list, so I don’t know what the other grand challenges are, which prevents contextualization. This doesn’t mean they’re making stuff up; for instance, Dill et al’s literature review of protein folding also says it’s a grand challenge, again without referring to a list of other challenges. How to square this? My non-expert take: it’s a grand challenge for computational biology, which while being “only” a subfield of biology still has huge real-world implications, so definitely worth celebrating; just takes a bit more explaining for fellow non-experts to appreciate.
[2] The “structure determines function” dogma is more complicated than it appears at first blush. For instance: Gregor Greslehner, a researcher at the University of Salzburg, published an interesting paper asking what molecular biologists really mean when they say “structure determines function”. Takeaways:
‘Structure’ and ‘function’ are ambiguous, referring to many different concepts
‘Structure → function’ is false anyway in the sense that evolution is pragmatic: selection works not on structures but functions -- that structures get reused for novel functions is a truism in evolutionary biology
The → relation isn’t determination but supervenience: 3D protein structure supervenes on their biochemical activities (X supervenes on Y iff a difference in Y is needed for a difference in X, but not necessarily the other way round)
My non-expert take: this nuance is a philosophical one; it doesn’t matter for DeepMind. The other criticisms above are more relevant.
[3] We do have a rough idea of what makes linear sequences spontaneously fold into globular proteins. For instance, some amino acids are hydrophobic, so they collapse towards the center to become shielded from the aqueous environment, a process called hydrophobic collapse:
(Source)
{kind=link}
Besides hydrophobic interactions, protein folding is guided by van der Waals forces, formation of intramolecular hydrogen bonds and so on.
[4] The basic idea in x-ray crystallography is to beam x-rays through proteins placed in crystals, whose electron clouds surrounding individual atoms diffract the beams in precise ways to create unique diffraction patterns. The patterns are then “read” by relating the x-ray amplitudes to the electron density clouds, letting us reconstruct the protein’s 3D structure:

(Source)
{kind=link}
[5] Why are trans-membrane proteins hard to crystallize? Because placing them in crystals entails taking them out of their cell membrane environment. And you can’t do that:
In water, charged parts will tend to rotate outward. In neutrally charged hydrophobic environments, non-charged protein parts will try to face outward. Cell membrane is a partially-hydrophobic environment, so one of the standard protein-folding rules is inverted in its fatty-layer zone. When taken out of it, the protein may literally flip (and also clump, with both itself and other membrane proteins). And since a sizable fraction of [trans-membrane] proteins are cross-membrane pores (transporting a specific molecule from one side of the membrane to the other), getting that cross-membrane portion accurately matters a LOT if you're trying to understand function.
[6] In a bit more detail -- predictions are scored in the Global Distance Test as the sum of scores of positional accuracies of individual atoms in the protein according to the following:
Award ¼ point if it’s within 0.8 nm of the experimentally-determined position
Award ½ point if it’s within 0.4 nm
Award ¾ point if it’s within 0.2 nm
Award 1 point if it’s within 0.1 nm
Mathematically, getting a 92.4 GDT like AlphaFold 2 did requires at least 85% of the structure prediction to be within 0.2 nm. For context, the carbon atoms that make up amino acids in proteins are ~0.15 nm across. The 90 GDT requirement is stringent.
[7] Mo AlQuraishi chastised DeepMind for not sharing details (emphasis mine):
I have to do something which I very much dislike to do but feel that I must—call out DeepMind for falling short of the standards of academic communication. What was presented on AF2 at CASP14 barely resembled a methods talk. It was exceedingly high-level, heavy on ideas and insinuations but almost entirely devoid of detail. This is a shame and contrasts markedly with DeepMind’s participation in CASP13, when they gave two talks that provided sufficient details for many groups to reproduce their results right away and participated in a poster session where they freely answered questions and built rapport with the community. While at CASP13 I and many others were surprised by DM’s entry and impressed by their results, we all walked away feeling like there’s a great new group of colleagues in the community. I’m afraid this time they left a different impression and I’m not sure it was at all necessary. DeepMind is in an exceedingly dominant position here—they will invariably get the cover of Nature or Science and may one day nab their first Nobel prize for AF2. Withholding details stands to poison the well of goodwill in the community. I hope their paper corrects it, and I furthermore hope they preprint their results to accelerate dissemination of their work.
[8] Mohammed AlQuraishi again (emphasis mine):
There is one other aspect of AF2 worth commenting on that doesn’t have to do with the approach per se but is interesting in its own right and is something of a mystery to me: the compute resources used. For training, AF2 consumed something like 128 TPUs for several weeks. That’s a lot of compute by academic standards but is not surprising for this type of problem. What is surprising however is the amount of compute needed for inference, i.e., making new predictions. According to Demis Hassabis, depending on the protein, they used between 5 to 40 GPUs for hours to days. Although they phrased this as “moderate” it is anything but for inference purposes. It is in fact an insane amount given that they are not doing any MD and has me perplexed. Nothing in their architecture as I understand it could warrant this much compute, unless they’re initializing from multiple random seeds for each prediction. The most computationally intensive part is likely the iterative MSA/distogram attention ping-pong, but even if that is run for hundreds or thousands of iterations, the inference compute seems too much. MSAs can be very large, that is true, but I doubt that they’re using them in their entirety as that seems overkill. At any rate I wonder if there is something to be learned from this mystery or if I am missing something obvious.
[9] Mohammed AlQuraishi yet again -- just a super-informative post (emphasis mine):
I would like to focus on organizational structure as I believe it is the key factor beyond the individual contributors themselves. DeepMind is organized very differently from academic groups. There are minimal administrative requirements, freeing up time to do research. This research is done by professionals working at the same job for years and who have achieved mastery of at least one discipline. Contrast this with academic labs where there is constant turnover of students and postdocs. This is as it should be, as their primary mission is the training of the next generation of scientists. Furthermore, at DeepMind everyone is rowing in the same direction. There is a reason that the AF2 abstract has 18 co-first authors and it is reflective of an incentive structure wholly foreign to academia. Research at universities is ultimately about individual effort and building a personal brand, irrespective of how collaborative one wants to be. This means the power of coordination that DeepMind can leverage is never available to academic groups. Taken together these factors result in a “fast and focused” research paradigm.
[10] Mohammed AlQuraishi on why you wouldn’t want to turn academia into “many mini DeepMinds” (emphasis mine):
To be clear, the DeepMind approach is no silver bullet. The factors I mentioned above—experienced hands, high coordination, and focused research objectives—are great for answering questions but not for asking them, whereas in most of biology defining questions is the interesting part; protein structure prediction being one major counterexample. It would be short-sighted to turn the entire research enterprise into many mini DeepMinds.
There is another, more subtle drawback to the fast and focused model and that is its speed. Even for protein structure prediction, if DeepMind’s research had been carried out over a period of ten years instead of four, it is likely that their ideas, as well as other ideas they didn’t conceive of, would have slowly gestated and gotten published by multiple labs. Some of these ideas may or may not have ultimately contributed to the solution, but they would have formed an intellectual corpus that informs problems beyond protein structure prediction. The fast and focused model minimizes the percolation and exploration of ideas. Instead of a thousand flowers blooming, only one will, and it may prevent future bloomings by stripping them of perceived academic novelty. Worsening matters is that while DeepMind may have tried many approaches internally, we will only hear about a single distilled and beautified result.
None of this is DeepMind’s fault—it reflects the academic incentive structure, particularly in biology (and machine learning) that elevates bottom-line performance over the exploration of new ideas. This is what I mean by stripping them from perceived academic novelty. Once a solution is solved in any way, it becomes hard to justify solving it another way, especially from a publication standpoint.”
[11] Carlos Rubiera on how DeepMind leveraged the decades-long output of researchers:
A major piece of DeepMind’s success is the availability of techniques, and especially data that has been painstakingly collected by structural biology groups for decades. The Protein Data Bank, which they used for training, accounts for ~170,000 structures, most of them produced by academic groups, as happens with the UniRef protein database, or the BFD and MGnify clusters of metagenomics sequences. The software tools employed, like HHblits, JackHMMER and OpenMM were also developed by government-funded academic initiatives. Also important — most of these initiatives were funded with public money. Big as DeepMind’s war chest might be, the taxpayers’ investment that has made their achievement possible is several orders of magnitude larger.
This is no less true for the wide body of research about protein structure prediction that is available in peer-reviewed articles, which has been conducted, written and reviewed by academics. This includes many of the ideas that AlphaFold incorporates, from exploiting a multiple sequence alignment to predict protein structure, to incorporating templates into modelling. This is intending in no way to diminish DeepMind’s work. They have developed a novel way of tackling protein structure prediction which combines many creative ideas with superb engineering. But, if they have managed to see further, it is because they stood on the shoulders of giants.